

作者:神经内科 黄刚
奔走跑跳,是人类天生的本能。然而,有些人走路晚、走路慢,容易摔倒。他们一生中都要和这种罕见病做斗争。近日,漯河市中心医院收治了一例主因“双下肢行走发软、易跌倒20年,加重3年”的32岁男性。经过神经内科、肌电图室、病理科、磁共振科等多学科会诊,完善心肌酶学、肌电图、肌肉活检和病理、肌肉磁共振、基因检测及家系分析等检查后,该患者最终被确诊为Becker型肌营养不良。在进行多器官系统全面综合评估后,制定合理治疗方案和随访计划,患者及家属满意出院。
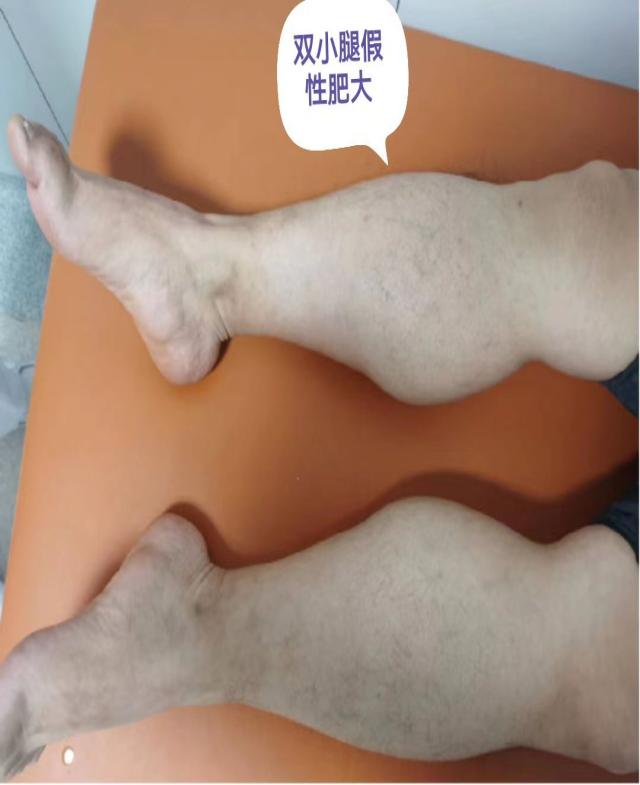

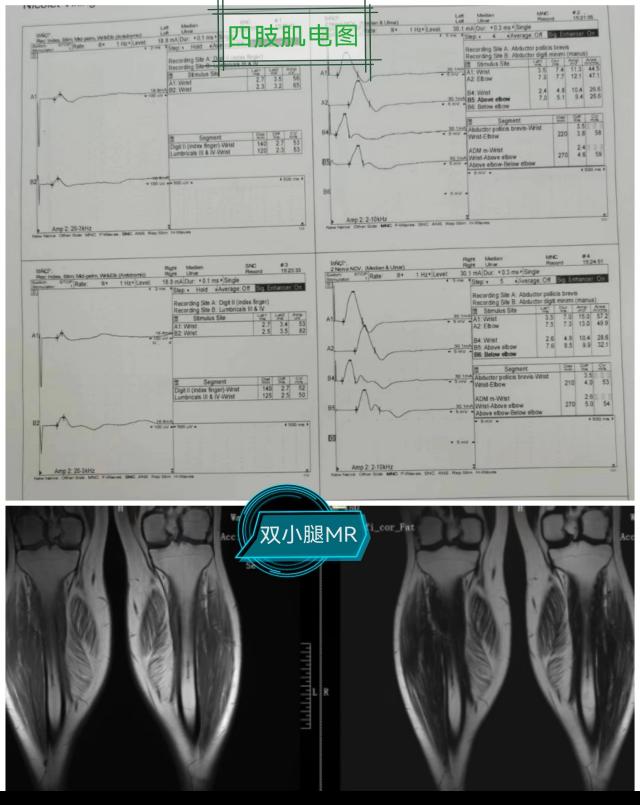
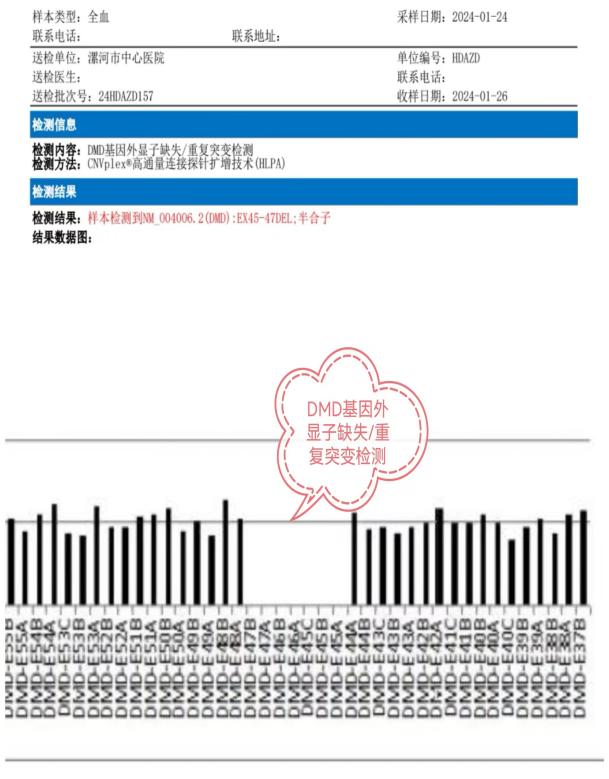
该例罕见病的确诊在彰显中心医院诊治神经疑难病症多学科协助综合实力的同时,也有力地促进了神经内科(周围神经病)亚专科的建设,并提高了综合医院的教学水平。接下来,就一起认识一下,什么是假肥大型肌营养不良吧!
1 概念
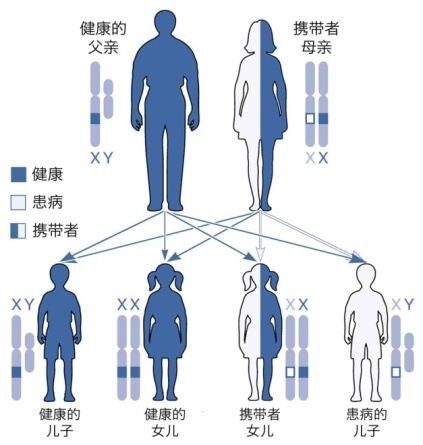
假肥大性肌营养不良,包括Duchene型肌营养不良症(DMD)和Becker型肌营养不良症(BMD),是全球最常见也是最严重的儿童遗传病之一。DMD呈X连锁隐性遗传,因此,绝大多数患者都是男性。主要临床表现为进行性加重的四肢近端肌、腰带肌无力、萎缩,腓肠肌肥大,严重影响患者的日常运动能力,病程晚期可累及呼吸肌和心肌。
它发病率为1/3500,我国每年约有400-500例DMD患儿出生,累计约7万人确诊为DMD。

‎
2 发病机制
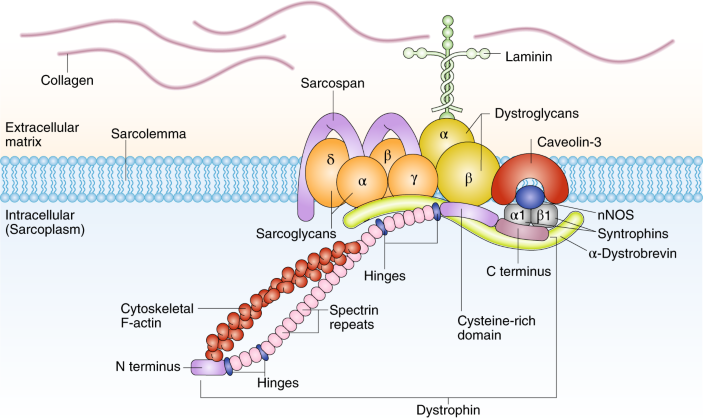
DMD的致病基因为抗肌萎缩蛋白基因(dystrophin),位于染色体Xp21.2区,全长约2.2Mb,共包含79个外显子,是已知最大的人类基因。抗肌萎缩蛋白主要位于骨骼肌和心肌细胞膜的质膜面,具有细胞支架、抗牵拉、防止肌细胞膜在收缩活动时撕裂的功能。
DMD 患者因基因缺陷而使肌细胞内缺乏抗肌萎缩蛋白,造成肌细胞膜不稳定并导致肌细胞坏死和功能缺失而发病,肌纤维的完整性遭到破坏,严重者可见纤维结缔组织和脂肪组织替代正常的肌肉组织,出现肌肉假性肥大的典型表现。
3 临床特征
(1)Duchenne型肌营养不良
幼儿期运动发育轻度迟滞,儿童期(5~6岁)运动能力开始下降,并出现步态异常、跟腱挛缩、腰椎前凸等变化。骨骼肌进行性无力萎缩,影响肢体运动功能,逐渐出现步态异常、上肢活动受限,自然病程常在10岁左右丧失行走能力。长期随访显示,DMD患者大多数活不过20岁,就会因心肺功能衰竭而死。
(2)Becker型肌营养不良
为同一疾病的相对良性表型,发病率为DMD的1/10。因DMD基因功能未完全丧失,所以病情明显轻于Duchenne型肌营养不良。可青年甚至成年起病,假肥大体征明显,基本不影响生存期,智力正常。

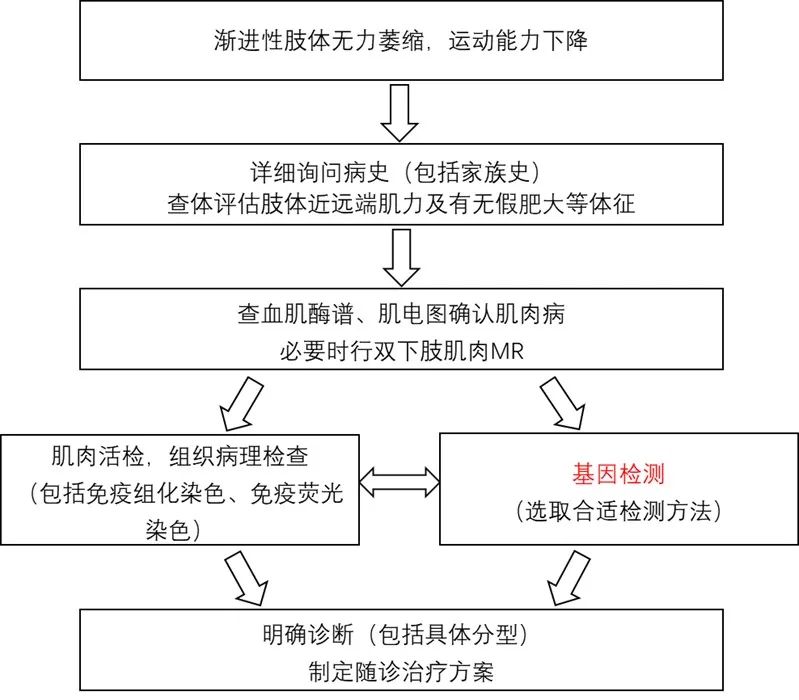
4 辅助检查和诊治流程
DMD和BMD的诊断相关辅助检查包括:血清学检测、肌电图、肌肉MRI、肌肉活检和基因检测,确诊需基因检测发现DMD基因致病性缺陷或肌肉活检发现Dystrophin蛋白异常。

5 治疗
迄今为止,DMD尚无有效的根治方法,但是及时的对症支持治疗和恰当的护理可以提高患者的生活质量和延长寿命。应鼓励患者尽可能从事日常活动,避免长期卧床。药物可选用小剂量激素、ATP、肌苷、维生素E等。
DMD的治疗策略的研发是与疾病赛跑的过程,对于肌营养不良这种疾病来说可以采用基因疗法进行治疗,而致病基因鉴定是实现治疗的第一步,通过基因检测明确致病基因,针对性的防治,帮助后代不再患病!基因治疗(外显子跳跃、微小基因替代)及干细胞移植治疗改变了这些曾经无法治愈的遗传疾病的治疗前景,为有意义的治疗干预带来新的希望!









